The PySPH framework¶
This document is an introduction to the design of PySPH. This provides additional high-level details on the functionality that the PySPH framework provides. This should allow you to use PySPH effectively and extend the framework to solve problems other than those provided in the main distribution.
To elucidate some of the internal details of PySPH, we will consider a typical SPH problem and proceed to write the code that implements it. Thereafter, we will look at how this is implemented using the PySPH framework.
The dam-break problem¶
The problem that is used for the illustration is the Weakly Compressible SPH (WCSPH) formulation for free surface flows, applied to a breaking dam problem:
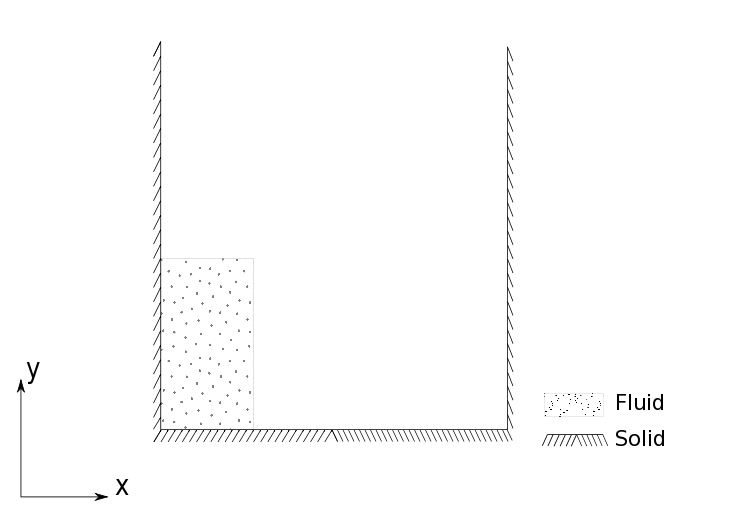
A column of water is initially at rest (presumably held in place by some membrane). The problem simulates a breaking dam in that the membrane is instantly removed and the column is free to fall under its own weight and the effect of gravity. This and other variants of the dam break problem can be found in the examples directory of PySPH.
Equations¶
The discrete equations for this formulation are given as
Boundary conditions¶
The dam break problem involves two types of particles. Namely, the fluid (water column) and solid (tank). The basic boundary condition enforced on a solid wall is the no-penetration boundary condition which can be stated as
Where \(\vec{n_b}\) is the local normal vector for the boundary. For this example, we use the dynamic boundary conditions. For this boundary condition, the boundary particles are treated as fixed fluid particles that evolve with the continuity ((2)) and equation the of state ((1)). In addition, they contribute to the fluid acceleration via the momentum equation ((3)). When fluid particles approach a solid wall, the density of the fluids and the solids increase via the continuity equation. With the increased density and consequently increased pressure, the boundary particles express a repulsive force on the fluid particles, thereby enforcing the no-penetration condition.
Time integration¶
For the time integration, we use a second order predictor-corrector integrator. For the predictor stage, the following operations are carried out:
Once the variables are predicted to their half time step values, the pairwise interactions are carried out to compute the accelerations. Subsequently, the corrector is used to update the particle positions:
Note
The acceleration variables are prefixed like \(a_\). The boldface symbols in the above equations indicate vector quantities. Thus \(a_\boldsymbol{v}\) represents \(a_u,\, a_v,\, \text{and}\, a_w\) for the vector components of acceleration.
Required arrays and properties¶
We will be using two ParticleArrays (see
pysph.base.particle_array.ParticleArray), one for the fluid and
another for the solid. Recall that for the dynamic boundary conditions, the
solid is treated like a fluid with the only difference being that the velocity
(\(a_\boldsymbol{v}\)) and position accelerations (\(a_\boldsymbol{x}
= \boldsymbol{u} + \boldsymbol{u}^{\text{XSPH}}\)) are never calculated. The
solid particles therefore remain fixed for the duration of the simulation.
To carry out the integrations for the particles, we require the following variables:
- SPH properties: x, y, z, u, v, w, h, m, rho, p, cs
- Acceleration variables: au, av, aw, ax, ay, az, arho
- Properties at the beginning of a time step: x0, y0, z0, u0, v0, w0, rho0
A non-PySPH implementation¶
We first consider the pseudo-code for the non-PySPH implementation. We assume
we have been given two ParticleArrays fluid and solid corresponding to
the dam-break problem. We also assume that an pysph.base.nnps.NNPS
object nps is available and can be used for neighbor queries:
from pysph.base import nnps
fluid = get_particle_array_fluid(...)
solid = get_particle_array_solid(...)
particles = [fluid, solid]
nps = nnps.LinkedListNNPS(dim=2, particles=particles, radius_scale=2.0)
The part of the code responsible for the interactions can be defined as
class SPHCalc:
def __init__(nnps, particles):
self.nnps = nnps
self.particles = particles
def compute(self):
self.eos()
self.accelerations()
def eos(self):
for array in self.particles:
num_particles = array.get_number_of_particles()
for i in range(num_particles):
array.p[i] = # TAIT EOS function for pressure
array.cs[i] = # TAIT EOS function for sound speed
def accelerations(self):
fluid, solid = self.particles[0], self.particles[1]
nps = self.nps
nbrs = UIntArray()
# continuity equation for the fluid
dst = fluid; dst_index = 0
# source is fluid
src = fluid; src_index = 0
num_particles = dst.get_number_of_particles()
for i in range(num_particles):
# get nearest fluid neigbors
nps.get_nearest_particles(src_index, dst_index, d_idx=i, nbrs)
for j in nbrs:
# pairwise quantities
xij = dst.x[i] - src.x[j]
yij = dst.y[i] - src.y[j]
...
# kernel interaction terms
wij = kenrel.function(xi, ...) # kernel function
dwij= kernel.gradient(xi, ...) # kernel gradient
# compute the interaction and store the contribution
dst.arho[i] += # interaction term
# source is solid
src = solid; src_index = 1
num_particles = dst.get_number_of_particles()
for i in range(num_particles):
# get nearest fluid neigbors
nps.get_nearest_particles(src_index, dst_index, d_idx=i, nbrs)
for j in nbrs:
# pairwise quantities
xij = dst.x[i] - src.x[j]
yij = dst.y[i] - src.y[j]
...
# kernel interaction terms
wij = kenrel.function(xi, ...) # kernel function
dwij= kernel.gradient(xi, ...) # kernel gradient
# compute the interaction and store the contribution
dst.arho[i] += # interaction term
# Destination is solid
dst = solid; dst_index = 1
# source is fluid
src = fluid; src_index = 0
num_particles = dst.get_number_of_particles()
for i in range(num_particles):
# get nearest fluid neigbors
nps.get_nearest_particles(src_index, dst_index, d_idx=i, nbrs)
for j in nbrs:
# pairwise quantities
xij = dst.x[i] - src.x[j]
yij = dst.y[i] - src.y[j]
...
# kernel interaction terms
wij = kenrel.function(xi, ...) # kernel function
dwij= kernel.gradient(xi, ...) # kernel gradient
# compute the interaction and store the contribution
dst.arho[i] += # interaction term
We see that the use of multiple particle arrays has forced us to write a fairly long piece of code for the accelerations. In fact, we have only shown the part of the main loop that computes \(a_\rho\) for the continuity equation. Recall that our problem states that the continuity equation should evaluated for all particles, taking influences from all other particles into account. For two particle arrays (fluid, solid), we have four such pairings (fluid-fluid, fluid-solid, solid-fluid, solid-solid). The last one can be eliminated when we consider the that the boundary has zero velocity and hence the contribution will always be trivially zero.
The apparent complexity of the SPHCalc.accelerations method notwithstanding, we notice that similar pieces of the code are being repeated. In general, we can break down the computation for a general source-destination pair like so:
# consider first destination particle array
for all dst particles:
get_neighbors_from_source()
for all neighbors:
compute_pairwise_terms()
compute_inteactions_for_dst_particle()
# consider next source for this destination particle array
...
# consider the next destination particle array
Note
The SPHCalc.compute method first calls the EOS before calling the main loop to compute the accelerations. This is because the EOS (which updates the pressure) must logically be completed for all particles before the accelerations (which uses the pressure) are computed.
The predictor-corrector integrator for this problem can be defined as
class Integrator:
def __init__(self, particles, nps, calc):
self.particles = particles
self.nps = nps
self.calc = calc
def initialize(self):
for array in self.particles:
array.rho0[:] = array.rho[:]
...
array.w0[:] = array.w[:]
def stage1(self, dt):
dtb2 = 0.5 * dt
for array in self.particles:
array.rho = array.rho0[:] + dtb2*array.arho[:]
array.u = array.u0[:] + dtb2*array.au[:]
array.v = array.v0[:] + dtb2*array.av[:]
...
array.z = array.z0[:] + dtb2*array.az[:]
def stage2(self, dt):
for array in self.particles:
array.rho = array.rho0[:] + dt*array.arho[:]
array.u = array.u0[:] + dt*array.au[:]
array.v = array.v0[:] + dt*array.av[:]
...
array.z = array.z0[:] + dt*array.az[:]
def integrate(self, dt):
self.initialize()
self.stage1(dt) # predictor step
self.nps.update() # update NNPS structure
self.calc.compute() # compute the accelerations
self.stage2(dt) # corrector step
The Integrator.integrate method is responsible for updating the solution the next time level. Before the predictor stage, the Integrator.initialize method is called to store the values x0, y0… at the beginning of a time-step. Given the positions of the particles at the half time-step, the NNPS data structure is updated before calling the SPHCalc.compute method. Finally, the corrector step is called once we have the updated accelerations.
This hypothetical implementation can be integrated to the final time by calling the Integrator.integrate method repeatedly. In the next section, we will see how PySPH does this automatically.
PySPH implementation¶
Now that we have a hypothetical implementation outlined, we can proceed to describe the abstractions that PySPH introduces, enabling a highly user friendly and flexible way to define pairwise particle interactions. To see a working example, see dam_break_2d.py.
We assume that we have the same ParticleArrays (fluid and solid) and NNPS objects as before.
Specifying the equations¶
Given the particle arrays, we ask for a given set of operations to be
performed on the particles by passing a list of Equation objects (see
SPH equations) to the Solver (see
pysph.solver.solver.Solver)
equations = [
# Equation of state
Group(equations=[
TaitEOS(dest='fluid', sources=None, rho0=ro, c0=co, gamma=gamma),
TaitEOS(dest='boundary', sources=None, rho0=ro, c0=co, gamma=gamma),
], real=False),
Group(equations=[
# Continuity equation
ContinuityEquation(dest='fluid', sources=['fluid', 'boundary']),
ContinuityEquation(dest='boundary', sources=['fluid']),
# Momentum equation
MomentumEquation(dest='fluid', sources=['fluid', 'boundary'],
alpha=alpha, beta=beta, gy=-9.81, c0=co),
# Position step with XSPH
XSPHCorrection(dest='fluid', sources=['fluid'])
]),
]
We see that we have used two Group objects (see
pysph.sph.equation.Group), segregating two parts of the evaluation
that are logically dependent. The second group, where the accelerations are
computed must be evaluated after the first group where the pressure is
updated. Recall we had to do a similar seggregation for the SPHCalc.compute
method in our hypothetical implementation:
class SPHCalc:
def __init__(nnps, particles):
...
def compute(self):
self.eos()
self.accelerations()
Note
PySPH will respect the order of the Equation and equation Groups as provided by the user. This flexibility also means it is quite easy to make subtle errors.
Note that in the first group, we have an additional parameter called
real=False. This is only relevant for parallel simulations and for
simulations with periodic boundaries. What it says is that the equations in
that group should be applied to all particles (remote and local), non-local
particles are not “real”. By default a Group has real=True, thus only
local particles are operated on. However, we wish to apply the Equation of
state on all particles. Similar is the case for periodic problems where it is
sometimes necessary to set real=True in order to set the properties of the
additional particles used for periodicity.
Writing the equations¶
It is important for users to be able to easily write out new SPH equations of motion. PySPH provides a very convenient way to write these equations. The PySPH framework allows the user to write these equations in pure Python. These pure Python equations are then used to generate high-performance code and then called appropriately to perform the simulations.
There are two types of particle computations in SPH simulations:
- The most common type of interaction is to change the property of one particle (the destination) using the properties of a source particle.
- A less common type of interaction is to calculate say a sum (or product or maximum or minimum) of values of a particular property. This is commonly called a “reduce” operation in the context of Map-reduce programming models.
Computations of the first kind are inherently parallel and easy to perform correctly both in serial and parallel. Computations of the second kind (reductions) can be tricky in parallel. As a result, in PySPH we distinguish between the two. This will be elaborated in more detail in the following.
In general an SPH algorithm proceeds as the following pseudo-code illustrates:
for destination in particles:
for equation in equations:
equation.initialize(destination)
# This is where bulk of the computation happens.
for destination in particles:
for source in destination.neighbors:
for equation in equations:
equation.loop(source, destination)
for destination in particles:
for equation in equations:
equation.post_loop(destination)
# Reduce any properties if needed.
total_mass = reduce_array(particles.m, 'sum')
max_u = reduce_array(particles.u, 'max')
The neighbors of a given particle are identified using a nearest neighbor algorithm. PySPH does this automatically for the user and internally uses a link-list based algorithm to identify neighbors.
In PySPH we follow some simple conventions when writing equations. Let us look
at a few equations first. In keeping the analogy with our hypothetical
implementation and the SPHCalc.accelerations method above, we consider the
implementations for the PySPH pysph.sph.wc.basic.TaitEOS and
pysph.sph.basic_equations.ContinuityEquation objects. The former
looks like:
class TaitEOS(Equation):
def __init__(self, dest, sources=None,
rho0=1000.0, c0=1.0, gamma=7.0):
self.rho0 = rho0
self.rho01 = 1.0/rho0
self.c0 = c0
self.gamma = gamma
self.gamma1 = 0.5*(gamma - 1.0)
self.B = rho0*c0*c0/gamma
super(TaitEOS, self).__init__(dest, sources)
def loop(self, d_idx, d_rho, d_p, d_cs):
ratio = d_rho[d_idx] * self.rho01
tmp = pow(ratio, self.gamma)
d_p[d_idx] = self.B * (tmp - 1.0)
d_cs[d_idx] = self.c0 * pow( ratio, self.gamma1 )
Notice that it has only one loop method and this loop is applied
for all particles. Since there are no sources, there is no need for
us to find the neighbors. There are a few important conventions that
are to be followed when writing the equations.
d_*indicates a destination array.s_*indicates a source array.d_idxands_idxrepresent the destination and source index respectively.- Each function can take any number of arguments as required, these are automatically supplied internally when the application runs.
- All the standard math symbols from
math.hare also available.
Let us look at the ContinuityEquation as another simple example.
It is instantiated as:
class ContinuityEquation(Equation):
def initialize(self, d_idx, d_arho):
d_arho[d_idx] = 0.0
def loop(self, d_idx, d_arho, s_idx, s_m, DWIJ, VIJ):
vijdotdwij = DWIJ[0]*VIJ[0] + DWIJ[1]*VIJ[1] + DWIJ[2]*VIJ[2]
d_arho[d_idx] += s_m[s_idx]*vijdotdwij
Notice that the initialize method merely sets the value to zero. The
loop method also accepts a few new quantities like DWIJ, VIJ etc.
These are precomputed quantities and are automatically provided depending on
the equations needed for a particular source/destination pair. The following
precomputed quantites are available and may be passed into any equation:
HIJ = 0.5*(d_h[d_idx] + s_h[s_idx]).XIJ[0] = d_x[d_idx] - s_x[s_idx],XIJ[1] = d_y[d_idx] - s_y[s_idx],XIJ[2] = d_z[d_idx] - s_z[s_idx]R2IJ = XIJ[0]*XIJ[0] + XIJ[1]*XIJ[1] + XIJ[2]*XIJ[2]RIJ = sqrt(R2IJ)WIJ = KERNEL(XIJ, RIJ, HIJ)WJ = KERNEL(XIJ, RIJ, s_h[s_idx])RHOIJ = 0.5*(d_rho[d_idx] + s_rho[s_idx])WI = KERNEL(XIJ, RIJ, d_h[d_idx])RHOIJ1 = 1.0/RHOIJDWIJ:GRADIENT(XIJ, RIJ, HIJ, DWIJ)DWJ:GRADIENT(XIJ, RIJ, s_h[s_idx], DWJ)DWI:GRADIENT(XIJ, RIJ, d_h[d_idx], DWI)VIJ[0] = d_u[d_idx] - s_u[s_idx]VIJ[1] = d_v[d_idx] - s_v[s_idx]VIJ[2] = d_w[d_idx] - s_w[s_idx]EPS = 0.01 * HIJ * HIJ
In addition if one requires the current time or the timestep in an equation, the following may be passed into any of the methods of an equation:
t: is the current time.dt: the current time step.
Note
Note that all standard functions and constants in math.h are available
for use in the equations. The value of \(\pi\) is available in
M_PI. Please avoid using functions from numpy as these are Python
functions and are slow. They also will not allow PySPH to be run with
OpenMP. Similarly, do not use functions or constants from sympy and
other libraries inside the equation methods as these will significantly
slow down your code.
In addition, these constants from the math library are available:
M_E: value of eM_LOG2E: value of log2eM_LOG10E: value of log10eM_LN2: value of loge2M_LN10: value of loge10M_PI: value of piM_PI_2: value of pi / 2M_PI_4: value of pi / 4M_1_PI: value of 1 / piM_2_PI: value of 2 / piM_2_SQRTPI: value of 2 / (square root of pi)M_SQRT2: value of square root of 2M_SQRT1_2: value of square root of 1/2
In an equation, any undeclared variables are automatically declared to be
doubles in the high-performance Cython code that is generated. In addition
one may declare a temporary variable to be a matrix or a cPoint by
writing:
mat = declare("matrix((3,3))")
point = declare("cPoint")
When the Cython code is generated, this gets translated to:
cdef double[3][3] mat
cdef cPoint point
One can also declare any valid c-type using the same approach, for example if
one desires a long data type, one may use ii = declare("long").
One may also perform any reductions on properties. Consider a trivial example
of calculating the total mass and the maximum u velocity in the following
equation:
class FindMaxU(Equation):
def reduce(self, dst, t, dt):
m = serial_reduce_array(dst.m, 'sum')
max_u = serial_reduce_array(dst.u, 'max')
dst.total_mass[0] = parallel_reduce_array(m, 'sum')
dst.max_u[0] = parallel_reduce_array(u, 'max')
where:
dst: refers to a destinationParticleArray.t, dt: are the current time and timestep respectively.serial_reduce_array: is a special function provided that performs reductions correctly in serial. It currently supportssum, prod, maxandminoperations. Seepysph.base.reduce_array.serial_reduce_array(). There is also apysph.base.reduce_array.parallel_reduce_array()which is to be used to reduce an array across processors. Usingparallel_reduce_arrayis expensive as it is an all-to-all communication. One can reduce these by using a single array and use that to reduce the communication.
We recommend that for any kind of reductions one always use the
serial_reduce_array function and the parallel_reduce_array inside a
reduce method. One should not worry about parallel/serial modes in this
case as this is automatically taken care of by the code generator. In serial,
the parallel reduction does nothing.
With this machinery, we are able to write complex equations to solve almost any SPH problem. A user can easily define a new equation and instantiate the equation in the list of equations to be passed to the application. It is often easiest to look at the many existing equations in PySPH and learn the general patterns.
If you wish to use adaptive time stepping, see the code
pysph.sph.integrator.Integrator. The integrator uses information
from the arrays dt_cfl, dt_force, and dt_visc in each of the
particle arrays to determine the most suitable time step.
For a more focused discussion on how you should write equations, please see Writing equations.
Writing the Integrator¶
The integrator stepper code is similar to the equations in that they are all written in pure Python and Cython code is automatically generated from it. The simplest integrator is the Euler integrator which looks like this:
class EulerIntegrator(Integrator):
def one_timestep(self, t, dt):
self.initialize()
self.compute_accelerations()
self.stage1()
self.do_post_stage(dt, 1)
Note that in this case the integrator only needs to implement one timestep
using the one_timestep method above. The initialize and stage
methods need to be implemented in stepper classes which perform the actual
stepping of the values. Here is the stepper for the Euler integrator:
class EulerStep(IntegratorStep):
def initialize(self):
pass
def stage1(self, d_idx, d_u, d_v, d_w, d_au, d_av, d_aw, d_x, d_y,
d_z, d_rho, d_arho, dt=0.0):
d_u[d_idx] += dt*d_au[d_idx]
d_v[d_idx] += dt*d_av[d_idx]
d_w[d_idx] += dt*d_aw[d_idx]
d_x[d_idx] += dt*d_u[d_idx]
d_y[d_idx] += dt*d_v[d_idx]
d_z[d_idx] += dt*d_w[d_idx]
d_rho[d_idx] += dt*d_arho[d_idx]
As can be seen the general structure is very similar to how equations are
written in that the functions take an arbitrary number of arguments and are
set. The value of dt is also provided automatically when the methods are
called.
It is important to note that if there are additional variables to be stepped in addition to these standard ones, you must write your own stepper. Currently, only certain steppers are supported by the framework. Take a look at the Integrator related modules for more examples.
Simulating periodicity¶
PySPH provides a simplistic implementation for problems with periodicity. The
pysph.base.nnps_base.DomainManager is used to specify this. To use
this in an application simply define a method as follows:
# ...
from pysph.base.nnps import DomainManager
class TaylorGreen(Application):
def create_domain(self):
return DomainManager(
xmin=0.0, xmax=1.0, ymin=0.0, ymax=1.0,
periodic_in_x=True, periodic_in_y=True
)
# ...
This is a 2D example but something similar can be done in 3D. How this works is that PySPH will automatically copy the appropriate layer of the particles from each side of the domain and create “Ghost” particles (these are not “real” particles). The properties of the particles will also be copied but this is done before any accelerations are computed. Note that this implies that the real particles should be created carefully so as to avoid two particles being placed at the same location.
For example in the above example, the domain is defined in the unit square
with one corner at the origin and the other at (1,1). If we place any
particles exactly at \(x=0.0\) they will be copied over to 1.0 and if we
place any particles at \(x=1.0\) they will be copied to \(x=0\). This
will mean that there will be one real particle at 0 and a copy from 1.0 as
well at the same location. It is therefore important to initialize the
particles starting at dx/2 and all the way up-to 1.0-dx/2 so as to get
a uniform distribution of particles without any repetitions. It is important
to remember that the periodic particles will be “ghost” particles and so any
equations that set properties like pressure should be in a group with
real=False.